Articolo di Franky

L’apprendimento automatico è stato applicato a molti problemi nel campo della cheminformatica e delle scienze biologiche, ad esempio per lo studio delle proprietà molecolari e lo sviluppo di nuovi farmaci. Una questione critica nella pipeline di risoluzione dei problemi per queste applicazioni è la selezione di una rappresentazione molecolare adeguata che caratterizzi il set di dati di destinazione e serva al modello a valle. La Figura 1 mostra un quadro concettuale per le diverse rappresentazioni molecolari. Di solito, una molecola è rappresentata in forma lineare, come una stringa SMILES, o in forma di grafo, come una matrice adiacente, magari con una matrice di attributi dei nodi per gli atomi e una matrice di attributi dei bordi per i legami. Una stringa SMILES può essere ulteriormente convertita in diversi formati, come l’impronta molecolare, la codifica one-hot o il word embedding. D’altra parte, la forma a grafo della rappresentazione molecolare potrebbe essere utilizzata direttamente dal modello a valle o essere convertita in embedding di grafo per il compito.
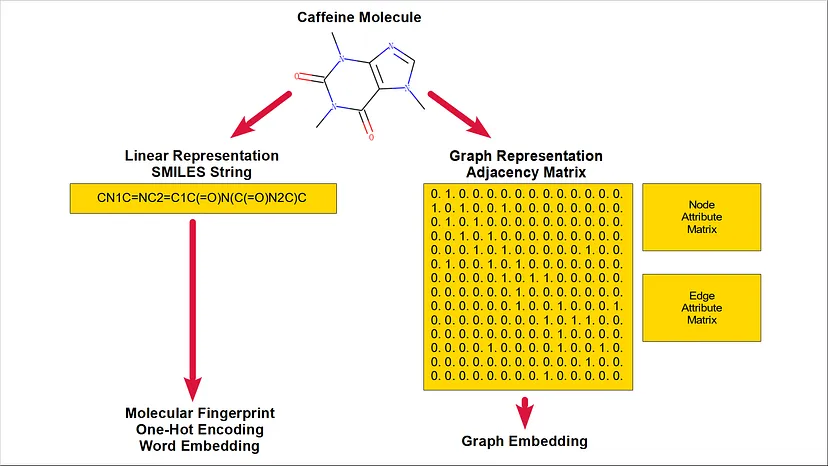
Questo post descrive alcuni codici utilizzati nell’implementazione del quadro concettuale di cui sopra, tra cui:
– lettura, disegno e analisi di una molecola,
– generazione dell’impronta molecolare da una stringa SMILES,
– la generazione della codifica one-hot da una stringa SMILES,
– generazione dell’incorporazione di parole da una stringa SMILES e
– generare una rappresentazione molecolare in un grafico
RDKit è una libreria open-source per la cheminformatica. Nel codice qua sotto viene mostrato come fare per la lettura della stringa SMILES della caffeina e il disegno della sua struttura molecolare. Si noti che C è il carbonio, N l’azoto e O l’ossigeno in una stringa SMILES.
# import RDKit ----------------------------------------------------------------
from rdkit import Chem
from rdkit.Chem import Draw
# define the smiles string and covert it into a molecule sturcture ------------
caffeine_smiles = 'CN1C=NC2=C1C(=O)N(C(=O)N2C)C'
mol = Chem.MolFromSmiles(caffeine_smiles)
# draw the modecule -----------------------------------------------------------
Draw.MolToFile(mol, 'caffeine.png')
# draw the molecule with property ---------------------------------------------
for i, atom in enumerate(mol.GetAtoms()):
atom.SetProp("molAtomMapNumber", str(atom.GetIdx()))
Draw.MolToFile(mol, 'caffeine_with_prop.png')
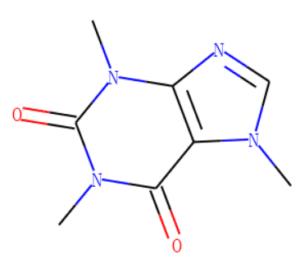
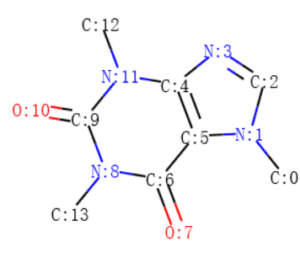
Una molecola può essere visualizzata senza l’etichettatura del carbonio, come mostrato nella Figura 2, o con l’etichettatura del carbonio, come mostrato nella Figura 3.
Il codice qua sotto, invece, mostra come visualizzare atomi e legami nella molecola della caffeina.
# import RDKit ---------------------------------------------------------------- from rdkit import Chem # define the smiles string and covert it into a molecule sturcture ------------ caffeine_smiles = 'CN1C=NC2=C1C(=O)N(C(=O)N2C)C' mol = Chem.MolFromSmiles(caffeine_smiles) # print the atoms of the molecule --------------------------------------------- for atom in mol.GetAtoms(): print(atom.GetIdx(),',', atom.GetAtomicNum(),',', atom.GetIsAromatic(),',', atom.GetSymbol()) # print the bonds of the molecule --------------------------------------------- for bond in mol.GetBonds(): print(bond.GetBeginAtomIdx(),',', bond.GetEndAtomIdx(),',', bond.GetBondType())
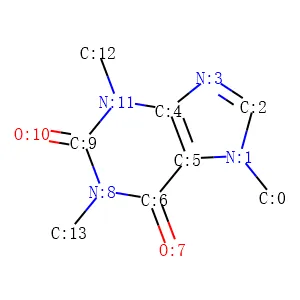
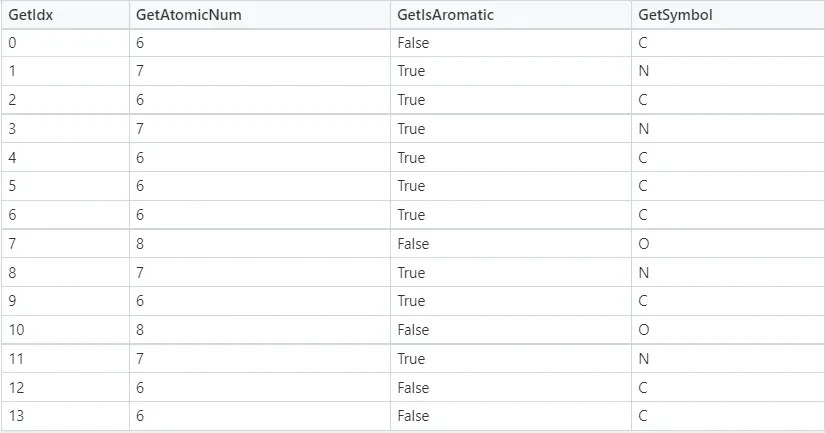
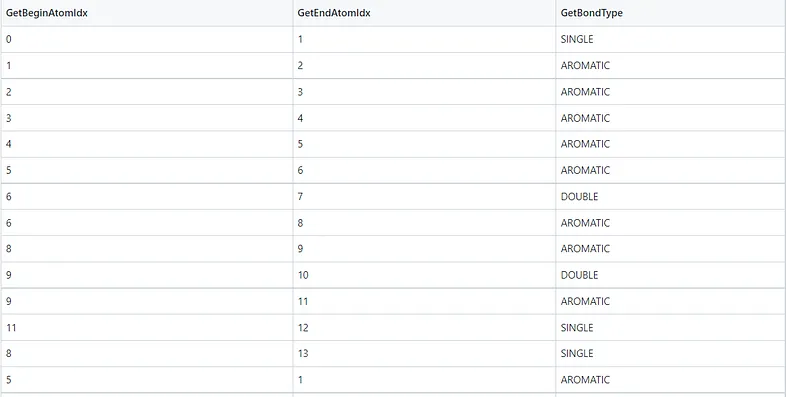
La Figura 4 e la Figura 5 mostrano rispettivamente i dettagli degli atomi e dei legami nella molecola della caffeina. Note:
1. Il termine “aromatico” potrebbe essere considerato semplicemente come “anello” nelle tabelle seguenti. GetIsAromatic nella Figura 4 indica se l’atomo è o meno in un anello e GetBondType nella Figura 5 indica se il legame è o meno in un anello.
2. Le Figure 4 e 5 possono essere considerate come una semplice matrice di attributi dell’atomo e una semplice matrice di attributi del legame nel quadro concettuale illustrato nella Figura 1.
3. L’elenco dei legami nella Figura 5 potrebbe rappresentare la forma del grafo di una molecola, cioè l’elenco dei legami di una matrice di adiacenza.
Nel contesto della modellazione linguistica, un approccio più sofisticato per generare una rappresentazione molecolare consiste nell’applicare il metodo del word embedding alle sottostrutture di una molecola. Il codice qua sotto mostra il processo di utilizzo di mol2vec e word2vec per generare il word embedding per tutte le molecole del dataset HIV.
# import rdkit/mol2vec/word2vec -----------------------------------------------
from rdkit import Chem
from mol2vec.features import mol2alt_sentence, MolSentence, DfVec, sentences2vec
from gensim.models import word2vec
# import numpy/pandas ---------------------------------------------------------
import numpy as np
import pandas as pd
print(">>> read the data file ... ")
hiv = pd.read_csv('HIV.csv')
print(">>> data shape = ", hiv.shape)
print(">>> data columns = ", hiv.columns, "\n")
print(hiv)
print()
print(">>> create mol from smiles ... ")
hiv['mol'] = hiv['smiles'].apply(lambda x: Chem.MolFromSmiles(x))
print(">>> create sentence from mol ... ")
hiv['sentence'] = hiv.apply(lambda x: MolSentence(mol2alt_sentence(x['mol'], radius=1)), axis=1)
print(">>> load the word2vec model ... ")
w2v_model = word2vec.Word2Vec.load('model_300dim.pkl')
print(">>> create embedding from sentence ... ")
hiv['embedding'] = [DfVec(x) for x in sentences2vec(hiv['sentence'], w2v_model)]
print(">>> data columns = ", hiv.columns, "\n")
hiv_mol2vec = np.array([x.vec for x in hiv['embedding']])
hiv_mol2vec = pd.DataFrame(hiv_mol2vec)
print(">>> hiv_mol2vec shape = ", hiv_mol2vec.shape)
print(hiv_mol2vec)
print()
Il dataset contiene 41127 molecole (Figura 6) e ogni molecola è codificata come un vettore a 300 dimensioni. Si noti che il codice è stato estratto da “Simple ML In Chemistry Rese.

Il processo di manipolazione di molecole/atomi/legami in RDKit fornisce le basi per la generazione di una rappresentazione molecolare in forma di grafico. Le Figure e 5, e il codice che le precede, hanno mostrato la matrice di adiacenza, la matrice degli attributi dei nodi e la rete degli attributi dei bordi della caffeina. Tuttavia, la conversione di una molecola in RDKit in un grafo in NetworkX (una libreria open-source per l’analisi delle reti) potrebbe sfruttare la ricerca degli algoritmi di grafi tradizionali e i moderni modelli di grafi per studiare la struttura e le proprietà molecolari. Il codice sottostante, invece, mostra la conversione di una molecola in RDKit in un grafo in NetworkX.
# import library --------------------------------------------------------------
from rdkit import Chem
import networkx as nx
import matplotlib.pyplot as plt
# define the smiles string and covert it into a molecule sturcture ------------
caffeine_smiles = 'CN1C=NC2=C1C(=O)N(C(=O)N2C)C'
caffeine_mol = Chem.MolFromSmiles(caffeine_smiles)
# define the function for coverting rdkit object to networkx object -----------
def mol_to_nx(mol):
G = nx.Graph()
for atom in mol.GetAtoms():
G.add_node(atom.GetIdx(),
atomic_num=atom.GetAtomicNum(),
is_aromatic=atom.GetIsAromatic(),
atom_symbol=atom.GetSymbol())
for bond in mol.GetBonds():
G.add_edge(bond.GetBeginAtomIdx(),
bond.GetEndAtomIdx(),
bond_type=bond.GetBondType())
return G
# conver rdkit object to networkx object --------------------------------------
caffeine_nx = mol_to_nx(caffeine_mol)
caffeine_atom = nx.get_node_attributes(caffeine_nx, 'atom_symbol')
color_map = {'C': 'cyan',
'O': 'orange',
'N': 'magenta'}
caffeine_colors = []
for idx in caffeine_nx.nodes():
if (caffeine_nx.nodes[idx]['atom_symbol'] in color_map):
caffeine_colors.append(color_map[caffeine_nx.nodes[idx]['atom_symbol']])
else:
caffeine_colors.append('gray')
nx.draw(caffeine_nx,
labels=caffeine_atom,
with_labels = True,
node_color=caffeine_colors,
node_size=800)
plt.show()
# print out the adjacency matrix ----------------------------------------------
matrix = nx.to_numpy_matrix(caffeine_nx)
print(matrix)
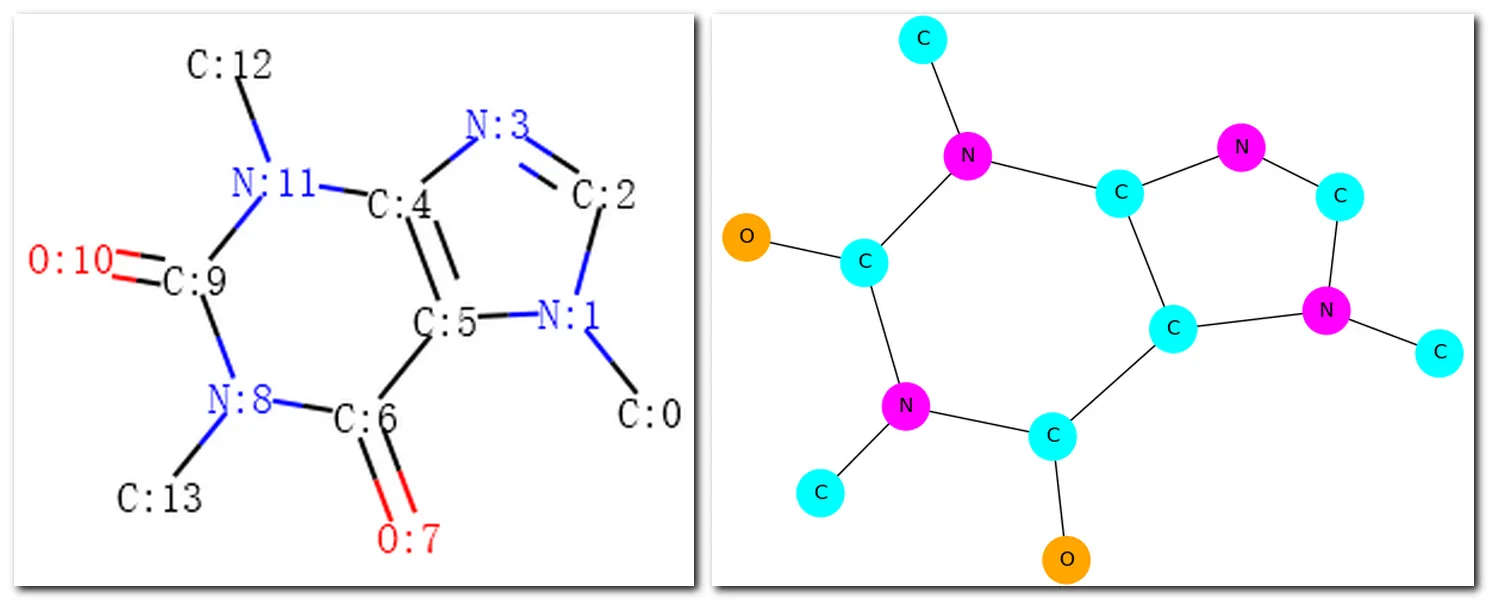
La Figura 7 mostra i grafi molecolari disegnati da RDKit e NetworkX.
Un’importante area di ricerca sulle reti di grafi è l’incorporazione dei grafi. In generale, l’embedding dei grafi si compone di tre argomenti: embedding a livello di nodo (che codifica i nodi di un grafo come vettori), embedding a livello di bordo (che codifica i bordi di un grafo come vettori) e embedding a livello di grafo (che codifica un intero grafo come vettore). Il codice qua sotto mostra come convertiree le molecole in RDKit in grafi in NetworkX e generare i relativi incorporamenti di grafi tramite Graph2Vec in KarateClub. Graph2Vec è un algoritmo di incorporamento dei grafi e KarateClub è un pacchetto che fornisce modelli di apprendimento automatico non supervisionato per i dati dei grafi.
# import rdkit/networkx/graph2vec ---------------------------------------------
from rdkit import Chem
import networkx as nx
from karateclub import Graph2Vec
# import numpy/pandas ---------------------------------------------------------
import numpy as np
import pandas as pd
print(">>> read the data file ... ")
hiv = pd.read_csv('HIV.csv')
print(">>> data shape = ", hiv.shape)
print(">>> data columns = ", hiv.columns, "\n")
print(hiv)
print()
print(">>> create mol from smiles ... ")
hiv['mol'] = hiv['smiles'].apply(lambda x: Chem.MolFromSmiles(x))
# define the function for coverting rdkit object to networkx object -----------
def mol_to_nx(mol):
G = nx.Graph()
for atom in mol.GetAtoms():
G.add_node(atom.GetIdx(),
atomic_num=atom.GetAtomicNum(),
is_aromatic=atom.GetIsAromatic(),
atom_symbol=atom.GetSymbol())
for bond in mol.GetBonds():
G.add_edge(bond.GetBeginAtomIdx(),
bond.GetEndAtomIdx(),
bond_type=bond.GetBondType())
return G
print(">>> create nx from mol ... ")
hiv['graph'] = hiv['mol'].apply(lambda x: mol_to_nx(x))
print(">>> create graph embedding ... ")
model = Graph2Vec()
model.fit(hiv['graph'])
hiv_graph2vec = model.get_embedding()
hiv_graph2vec = pd.DataFrame(hiv_graph2vec)
print(">>> hiv_graph2vec shape = ", hiv_graph2vec.shape)
print(hiv_graph2vec)
print()
