Articolo in lingua originale in lingua inglese di Anuradha Wickramarachchi
In generale, gli autoencoder mirano ad imparare una rappresentazione dei dati in una dimensione inferiore rispetto a quella di partenza.Uno dei maggiori vantaggi degli autoencoder è quello di essere in grado di imparare dimensioni significativamente piccole al contrario delle decomposizioni simili alla PCA che sono limitate dalla loro natura lineare. Sentitevi liberi di dare un’occhiata anche il mio articolo riguardo gli autoencoders.

Come al solito, parlerò di un’applicazione nell’ambito della bioinformatica
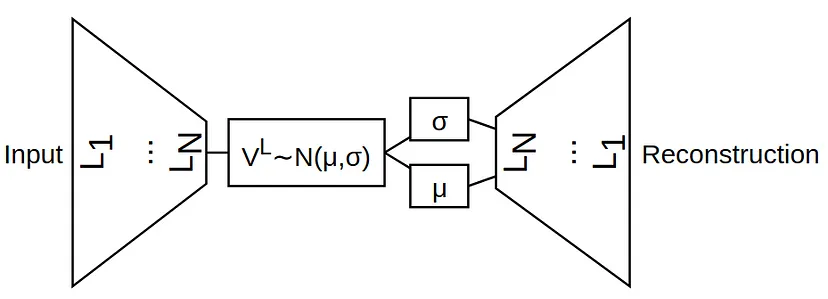
Simile ad un AE, c’è una regione a collo di bottiglia seguita da una ricostruzione. Più formalmente, abbiamo un encoder e un decoder. Da notare la rappresentazione latente VL e le successive variabili mu e sigma. La ricostruzione è generata dalle distribuzioni di questi parametri. La classe completa del VAE viene rappresentata come segue:

Si noti che viene utilizzata la funzione di attivazione RELU in molte occasioni. Tuttavia, nel caso della variabile latente logvar, viene scelto softplus. Questo perchè la varianza logaritmica è sempre positiva. Nella ricostruzione finale si utilizza una sigmoide dal momento che le dimensioni dei dati in input variano tra 0 e 1.
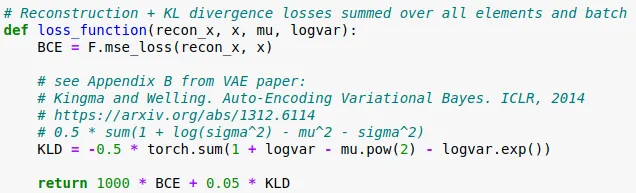
La funzione di perdita è costituita da due parti. La ricostruzione della perdita (RL) e la divergenza-KL (KLD). In particolare si utilizza KLD per assicurare che le distribuzioni apprese dalla rete siano il più vicine possibile ad una distribuzione normale (o Gaussiana, o similari). Potete leggere di più qui.
La ricostruzione è un buon vecchio Errore Quadratico Medio. In base all’applicazione, però, questo potrebbe cambiare. Ad esempio, per immagini in bianco e nero (MNIST) si userebbe la Binary Crossentropy.
Infine, possiamo cercare iperparametri ad-hoc per i pesi della RL e della KLD in modo che permettano il miglior clustering (binning o altro).
Consideriamo ora il seguente dataset di metagenomica da uno dei miei paper più recenti. Le reads più lunghe sono state simulate utilizzando l’applicazione SimLoRD.
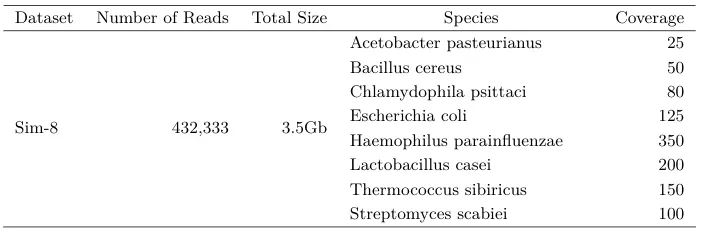
Ora il dataset è stato vettorizzato utilizzando il tool: https://github.com/anuradhawick/seq2vec, inteso
per la generazione dei dati nel Machine Learning in bioinformatica.
Il caricamento dei dati può essere realizzato come segue:

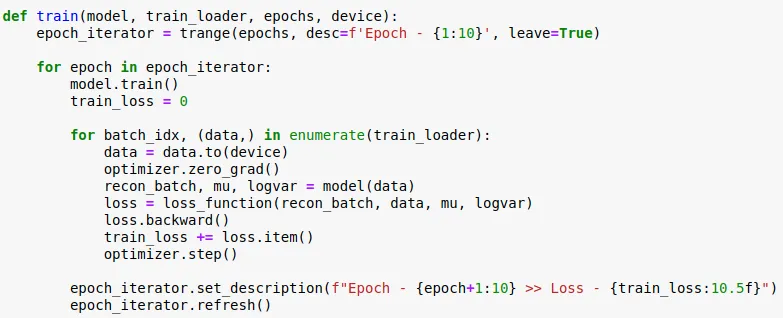
Mentre la funzione per addestrare il VAE:
L’inizializzazione
data = LOAD DATA truth = LOAD GROUND TRUTH # for visualizationdevice = "cuda" if torch.cuda.is_available() else "cpu"model = VAE(data.shape[1], 8).to(device) optimizer = optim.Adam(model.parameters(), lr=1e-2200)
Il training:
train_loader = make_data_loader(data, batch_size=1024, drop_last=True, shuffle=True, device=device)epochs = 50train(model, train_loader, epochs, device) epochs = 50train(model, train_loader, epochs, device)
Ottenere le rappresentazioni latenti:
with torch.no_grad(): model.eval() data = LOAD DATA data = torch.from_numpy(data).float().to(device) em, _ = model.encode(data)
Visualizzare:
import randomsidx = random.sample(range(len(data)), 10000) # just use 10000 em_2d = umap.UMAP().fit_transform(em.cpu().numpy()[sidx])plt.figure(figsize=(10, 10)) sns.scatterplot(x=em_2d.T[0], y=em_2d.T[1], hue=truth[sidx]) plt.legend(bbox_to_anchor=(1.05, 1))
Una volta addestrato il VAE si ottengono le rappresentazioni latenti. In questo esempio ho utilizzato UMAP per proiettare un campione di 10000 reads in uno spazio 2D per poterlo visualizzare. Si presentava cosi:

Guardando la figura possiamo vedere gruppi di punti di dati qui e là. Si può facilmente utilizzare uno strumento come HDBSCAN per estrarre i cluster più densi.
