
L’applicazione di Machine Learning e Deep Learning per la scoperta di farmaci, la genomica, la microscopia e la chimica quantistica può creare un impatto radicale e ha il potenziale per accelerare significativamente il processo di ricerca medica e sviluppo di vaccini, che è una necessità per qualsiasi pandemia come Covid19.
Prima ancora di iniziare, questo articolo è di altissimo livello e specificamente mirato per Data Scientist e ricercatori ML interessati alla scoperta di farmaci, specialmente durante il periodo di una pandemia esistente come Covid19. Se sei uno con un forte background in Bio-informatica o Chem-informatica e vuole avventurarsi nel mondo della scienza dei dati per questi casi d’uso, per favore contattami attraverso una delle opzioni menzionate qui, e possiamo discutere alcune opportunità interessanti per il bene superiore dell’umanità.
DeepChem, un framework open source, che utilizza internamente TensorFlow, che è stato specificamente progettato per semplificare la creazione di modelli di deep learning per varie applicazioni di scienze della vita.
In questo tutorial, vedremo come impostare DeepChem e vedremo come utilizzare DeepChem per:
- addestrare un modello in grado di prevedere la tossicità delle molecole
- addestramento di un modello per prevedere la solubilità delle molecole
- utilizzo di stringhe SMART per interrogare strutture molecolari.
SMARTS è un’estensione del linguaggio SMILES descritto in precedenza che può essere utilizzata per creare query.
# To gain a visual understanding of compounds in our dataset, let's draw them using rdkit. We define a couple of helper functions to get startedimport tempfile from rdkit import Chem from rdkit.Chem import Draw from itertools import islice from IPython.display import Image, displaydef display_images(filenames): """Helper to pretty-print images.""" for file in filenames: display(Image(file))def mols_to_pngs(mols, basename="test"): """Helper to write RDKit mols to png files.""" filenames = [] for i, mol in enumerate(mols): filename = "%s%d.png" % (basename, i) Draw.MolToFile(mol, filename) filenames.append(filename) return filenames
Ora, prendiamo una stringa SMILES di esempio e visualizziamo la struttura molecolare.
from rdkit import Chem from rdkit.Chem.Draw import MolsToGridImage smiles_list = ["CCCCC","CCOCC","CCNCC","CCSCC"] mol_list = [Chem.MolFromSmiles(x) for x in smiles_list] display_images(mols_to_pngs(mol_list))
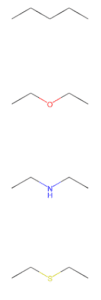
Questo è il modo in cui le strutture visive sono formate dalla stringa SMILES.
Ora, supponiamo di voler interrogare la stringa SMILES che ha tre carboni adiacenti.
query = Chem.MolFromSmarts("CCC")
match_list = [mol.GetSubstructMatch(query) for mol in
mol_list]
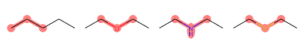
MolsToGridImage(mols=mol_list, molsPerRow=4,
highlightAtomLists=match_list)
![]()
Vediamo che la parte evidenziata rappresenta il composto con tre carboni adiacenti.
Allo stesso modo, vediamo alcune query di caratteri jolly e altre opzioni di query di sottostruttura.
query = Chem.MolFromSmarts("C*C")
match_list = [mol.GetSubstructMatch(query) for mol in
mol_list]
MolsToGridImage(mols=mol_list, molsPerRow=4,
highlightAtomLists=match_list)
query = Chem.MolFromSmarts("C[C,N,O]C")
match_list = [mol.GetSubstructMatch(query) for mol in
mol_list]
MolsToGridImage(mols=mol_list, molsPerRow=4,
highlightAtomLists=match_list)
![]()
Pertanto, possiamo vedere che anche la sottoquery selettiva può essere facilmente gestita.
Quindi, questo ci porta alla fine di questo articolo. So che questo articolo era di altissimo livello e specificamente mirato per Data Scientist e ricercatori ML interessati alla scoperta di farmaci, specialmente durante il periodo di una pandemia esistente come Covid19. Spero di essere stato in grado di aiutarti! Se sei una persona con un forte background in Bio-informatica o Chimica-informatica e vuoi avventurarti nel mondo della scienza dei dati, ti prego di contattarmi attraverso una delle opzioni menzionate qui. Continua a seguire: https://medium.com/@adib0073 e il mio sito web: https://www.aditya-bhattacharya.net/ per saperne di più!
Se, invece, siete interessati ad un corso che vi permetta di approfondire l’applicazione della Data Science in ambito biologico iscrivetevi al corso “Data Science e Bioinformatica” erogato da Francesco Lescai esclusivamente per Deep Learning Italia!
